 | ASE | 199 Members |
| Atomic Simulation Environment — https://wiki.fysik.dtu.dk/ase/ — Channel for discussions, support, and development of ASE | 3 Servers |
| ASE | 199 Members |
| Atomic Simulation Environment — https://wiki.fysik.dtu.dk/ase/ — Channel for discussions, support, and development of ASE | 3 Servers |
| Sender | Message | Time |
|---|---|---|
| 18 Jun 2024 | ||
 Toma Susi Toma Susi | yes so it does | 15:29:49 |
| Toma Susi | cool | 15:29:50 |
| Toma Susi | what we need additionally though is to pick specific phonon modee | 15:30:06 |
| Toma Susi | * what we need additionally though is to pick specific phonon modes | 15:30:10 |
| Toma Susi | the reason is that the multislice simulation doesn't know anything about energy loss, so we need to have the "correct phonon energy" in the structures | 15:30:34 |
 Florian Knoop Florian Knoop | In reply to @tomasusi:matrix.orgthere are ways to converge it but it's usually very fiddly https://github.com/tdep-developers/tdep-tutorials/tree/main/02_sampling/convergence/cutoff | 15:30:43 |
| Toma Susi | In reply to @tomasusi:matrix.orgit's a clever way of avoiding describing quite theoretically demanding inelastic momentum-resolved scattering | 15:31:31 |
| Florian Knoop | In reply to @tomasusi:matrix.orgI'd actually be interested to learn more about this | 15:31:57 |
| Florian Knoop | after spending a good deal of my postdoc on scattering theory (Raman) | 15:32:09 |
 Ask Hjorth Larsen Ask Hjorth Larsen | Andrew Rosen: Throughout most non-catastrophic periods in history we had about two releases per year, and things were eventually automatic enough to do more than that. I think once/month is exaggerated though, we have hardly changed anything. Once per 2-3 months ought to be quite fine. Let's get at least a few notable changes like plugins and ruff. | 15:38:33 |
| Toma Susi | In reply to @flokno:matrix.orgThat's the original paper: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.104.104301 | 15:49:01 |
| Florian Knoop | In reply to @tomasusi:matrix.orgthanks! | 15:57:41 |
 Hasith Vattikuti joined the room. Hasith Vattikuti joined the room. | 22:07:18 | |
| Hasith Vattikuti | When I view the dynamical matrix of a Phonons object, what exactly does the first index of D_N correspond to? I know that if we restrict ourselves to a 1x1x1 supercell, we have that each element is proportional to the partial of the potential of the system wrt the position of the i'th and j'th atom (as well as their masses) in the system. But how does a larger supercell change this? | 23:57:50 |
| 19 Jun 2024 | ||
 Anubhab Haldar Anubhab Haldar | The j'th atom can be in one of the other unit cells of the supercell. | 00:54:40 |
| Anubhab Haldar | I'm fairly certain N corresponds to the supercell counting index. | 00:54:57 |
| Anubhab Haldar | I'd look at the phonopy paper/docs which should have a reasonable exposition on this. | 00:55:21 |
| Ask Hjorth Larsen | Adam Jackson: Hi. Would you be able to continue review of the plugins MR? I can also do it later. | 11:14:40 |
 Adam Jackson Adam Jackson | Will try to take another look in the next couple of days | 11:42:24 |
| Hasith Vattikuti | In reply to @chronum:matrix.orgYeah, I also figured that in D_N[i,j,k], i corresponds to the supercell number, but I can't seem to find the page on phonopy docs about ordering the supercell components into an index, and the paper hardly mentions supercells at all. Do you know where I can find this? | 21:01:28 |
| 20 Jun 2024 | ||
| Anubhab Haldar | I don't, unfortunately. At that point you're likely better off looking at ASE's source code. | 00:35:29 |
| Hasith Vattikuti | In reply to @chronum:matrix.orgThanks for the suggestion, if anyone is curious, it looks like ASE orders the unit cells in the order of x,y,z | 05:18:04 |
| Hasith Vattikuti | Redacted or Malformed Event | 06:11:35 |
| Hasith Vattikuti | Redacted or Malformed Event | 06:11:36 |
| Hasith Vattikuti | Redacted or Malformed Event | 06:12:08 |
| Hasith Vattikuti | Can I get a little help with this? I attached my code and output, and can provide anything else, I can also make this a gitlab issue if that would be more appropriate. I set up three carbon atoms ~1.5 angstroms away from each other in a line on the x axis, and I expect the dynamical matrix to be of the form of a 9x9 matrix: [[2F, -F, 0], [-F, 2F, F], [0, -F, 2F]] where F=[[f, 0, 0], [0,0,0], [0,0,0]] (image attached). However, using the Phonons class, the dynamical matrix gives me only F--instead of 2F--in the upper left and lower right. The way I set up the phonon calculation is to use a 3x1x1 supercell of non-periodic unit cells. Could this have somehow caused the deviation from the theoretical calculation? Also there were some non-zero diagonal elements on F, which is strange since all the atoms were perfectly positioned along the x-axis (although they were usually only about a tenth of the value of f). Is this expected numerical error even though the setup was perfectly x-aligned? Code: Output: | 06:12:11 |
| Hasith Vattikuti | 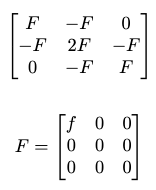 Download dynamical matrix.png | 06:13:27 |
 Robert Warmbier Robert Warmbier | PW mode doesn't understand non-periodic boundary conditions. | 07:34:25 |
| Robert Warmbier | And ase.phonon also seems to have periodic boundary conditions hard-coded. | 07:36:20 |
| Robert Warmbier | If you want your system to be non-periodic, you need to use ase.vibrations and/or add plenty of 'vacuum' left and right of your carbons | 07:37:31 |